


IMCAS ACADEMY | Webinar en español con la AEDV
22 junio, 2020
Juntos para hacer una gran dermatología en tiempos de coronavirus
26 junio, 2020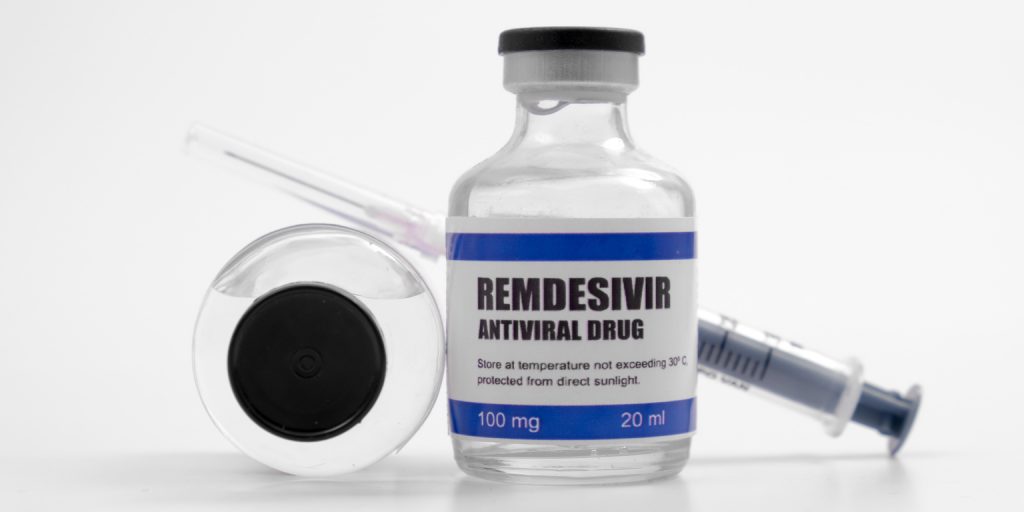
El comité de medicamentos humanos de la Agencia Europea del Medicamento (EMA, según sus siglas en inglés) recomienda otorgar la autorización de comercialización condicional a remdesivir para el tratamiento de adultos y adolescentes de 12 o más años con neumonía que requiere de oxigenoterapia provocada por la infección COVID-19.
Remdesivir es un fármaco antiviral que disminuye la carga viral del SARS-CoV-2. La medicación ya estaba utilizándose de forma compasiva y en ensayos clínicos en diferentes países, incluido el nuestro.
Con la nueva decisión, remdesivir pasa a ser el primer fármaco autorizado en la Unión Europea para el tratamiento de esta infección. Esta decisión viene respaldada por los datos evaluados en un plazo excepcionalmente corto y tras un procedimiento de revisión continua activado para emergencias de salud pública para evaluar datos a medida que están disponibles, según explica la Agencia en un comunicado que ha hecho público hace unas horas.
A través de este procedimiento, activado el 30 de abril, este comité ha evaluado datos sobre la calidad y fabricación, datos clínicos preliminares o datos de seguridad en programas de uso compasivo.
La recomendación de la EMA está basada, principalmente, en los datos del estudio NIAID-ACTT-1, patrocinado por el Instituto Nacional de Alergia y Enfermedades Infecciosas de Estados Unidos, junto con datos de otros estudios con remdesivir.
Pacientes que se benefician
Según los datos de estos trabajos, los pacientes graves tratados con este antiviral se recuperaron un promedio de un 31% más rápido que los que no reciben este fármaco. Sin embargo, esta ventaja no se ha detectado en personas con infección moderada o leve ni cuando el fármaco era administrado en pacientes que ya estaban con ventilación mecánica.
El medicamento es administrado por vía intravenosa, por lo que su uso está limitado al entorno hospitalario y vigilancia estrecha. La función hepática y renal deben de valorarse antes y durante el tratamiento y deben ser correctas. El tratamiento debe iniciarse con una infusión de 200mg el primer día, seguidos por otra de 100mg/día durante al menos 4 días y no más de 9 días.
La autorización condicional de la EMA significa que el fármaco puede comercializarse con menos datos y tiempo de los habituales, lo que facilita el acceso rápido al producto en el contexto de una pandemia y únicamente porque el beneficio de la disponibilidad inmediata supera a los posibles riesgos.
La empresa que desarrolla el fármaco, Gilead Sciences, deberá seguir presentando los datos de este fármaco en los próximos meses. Así, en agosto le mostrará al comité de la EMA la información completa sobre mortalidad y a finales de diciembre los informes finales de los estudios.
Está previsto que la Comisión Europea, en un procedimiento acelerado, apruebe este medicamento la próxima semana permitiendo así su comercialización en la UE bajo el nombre de Veklury durante un periodo de un año.



{kind=link}
{kind=link}